A knowledge modeling process suitable to satisfy these needs starts with identifying cues for classifying problem situations based on symbolic representations available as result of the automatic electrophoresis data preprocessing. The intricate aspect of this classification process is that it in the first place relies on results of electrophoresis analysis but will put these results under question as soon as the decision to tackle a certain problem has been made. Along with the decision to tackle a certain problem an inferencing goal has to be set up whether to argue in favour or in doubt of the hypothesis expressed as a result of the automatic electrophoresis. Support of the respective goal may come from sources as diverse as analog light absorption curves on the one side and second order structural properties of the DNA macromolecule and properties of the chemistry used to stain the strands. The resulting knowledge based support system is integrated into the workplace of the expert who is supposed to process those cases that the system passes. It is designed for optimal specifity i.e. in case of doubt will pass to the expert. This is to reflect the requirement of extremely high correctness - the tolerated error rate for the human - computer - integrated workplace is 0.03%. Nontheless the expectation is that by thorough guidance through human experts and their capability of identifying problem classes that are both common and relatively easy, a coverage of 30% of the doubtful cases can be achieved at an early stage.
Genetic information is coded in double strings (`helixes') each composed
of sequences of the four bases adenin, cytosin, guanin, and thymin (A,
C, G, and T). In the case of man, 23 double strings (`chromosomes') portray
the full genetic prodigy of a human being. The bases allow hydrogen bridge
bindings with specific complementary bases (adenin ![]() thymin, cytosin
thymin, cytosin ![]() guanin) each. As a consequence essential for propagating genetic information
both strands of the double helix have a blueprint character. Given the
sequence of bases found in one half, the complementary half is fully determined.
This essentially is the constraint or redundancy in the genetic code that
enables cell division and growth of offspring cells which replicate the
genetic code of the progenitor cell.
guanin) each. As a consequence essential for propagating genetic information
both strands of the double helix have a blueprint character. Given the
sequence of bases found in one half, the complementary half is fully determined.
This essentially is the constraint or redundancy in the genetic code that
enables cell division and growth of offspring cells which replicate the
genetic code of the progenitor cell.
Decoding the human (resp. other species) genetic information means
The mixture of these chains of different length can be seperated by gel electrophoresis to seperate the fragments by their different mobility and speed in an electric field. The bases pass a sensor in a single file and can be identified (read) one by one based on the light absorption of their respective stains by the base-caller.
Inhomogenities of the gel or of the electrophoresis procedure, impurifications, special properties of the used chemistry and secondary structures of the DNA result in unregular sensory signals and produce errors in the base calling procedure.
Because electrophoresis is able to read only sequences of a few hundred
bases with good quality (300 - 700) a lot of these reads (e.g. 600 to 1000
depending on the size of the cosmid etc.) of partially overlapping regions
are produced and sequenced. These sequences are fault tolerantly assembled
to larger sequences (called contigs) using the overlaps between them to
determine which reads to combine. Finding these overlaps is the fragment
assembly problem (Myers & Weber, 1997). Enough data is produced to
cover the cosmid about 4-6 fold on average. This redundancy of having a
couple of reads for each position uncovers base-calling errors, but repair
of an error cannot be decided based upon by a single majority vote. Because
of sparse data about some regions and errors in the electrophoresis, amplification
or base-calling, and assemply process we get more than a single contig
covering the whole segment (sometimes up to 100 or more) of interest. Thus
finding connections between contigs and misplaced reads in the contigs
is another editing problem.
![\includegraphics[width=12.5cm]{contig.eps}](img4.gif) |
Figure 1 displays an original overlay
of four differently coloured curves. Each colour denotes the intensity
detected for one of the bases. In sections where one curve displays a peak
dominating the intensities of the three other curves, the respective base
is called. In most non-obvious sections a heuristic call still takes
place, but the result is less reliable. Only in highly ambiguous sections
the base caller writes a dash (`-') instead of the letter of a base.
![\includegraphics[width=12.5cm]{traces.eps}](img5.gif) |
Figure 2 displays the sensory data from 3 aligned readings. Each column denotes one presumable site of a base. Each row denotes one reading. A letter A, C, G, or T in a row/column intersect denotes the base delivered by the base caller as the mostly likely one. An assessment of the certainty of the call is not supplied. Besides the dash for highly unclear sections we now encounter an asterisk (`*') as another special symbol. The asterisk is not written by the base caller but by the alignment algorithm. It denotes a site in one reading where no base had been suggested by the base caller, but where a base occurs in the respective sites of one or more other readings. It can be understood as the repair of an apparent leak in one reading as compared to parallelized readings.
The bottom line of the set of letter/dash/asterisks sequences inf figure 1 denotes the so called consensus. For each column i.e. without any context considerations it is defined as the symbol found in the majority of the lines, or a dash when there is no clear majority. The consensus is the basis of the final decision about the base that will be written into the world wide data bases of human genetics (e.g. GenBank or EMBL).
However, it can obviously not be used straight away. In the first place, the special symbols `*' and `-' have to be eliminated. In some cases a majority vote has to be outvoted based on a quality assessment of the original base readings. In extreme cases one dissenting call may become the consensus outvoting a clear majority. Presently, human experts check all columns with at least one special symbol or at least one dissenting call, search for possible joins between contigs and for possibly misplaced reads in contigs. This takes at least 2 or 3 days for a segment of cosmid size (40000 bases).
The revision of the consensus in all cases that are not absolutely unequivocal is among the tasks to be fulfilled by the knowledge based system (KBS). Further tasks will be outlined during the description of the model being developed. As to consensus revision, different problems occur according to presence/absence of asterisks or dashes. Different problems require to define different goals to be proven. But in the end, all inferences have to draw upon the original sensor data from gel electrophoresis. Some inferences do with assessments of individual readings. Complex problems require comparative assessments of parallel readings. More complex problems furthermore require protocol related features of readings. The knowledge engineering process has to be conducted in such a way that some solutions for certain problems become fully and reliably operational early. These early partial solutions should, however, model those inferences that occur again as part of methods for more complex problems, in a reusable form.
On the highest level of structuring, an expert chooses or becomes in charge of an overall task. This task allocation - in contrast to all further structures to follow below - is fully goal or process management driven. One task that is already obvious from the outline of the domain is to clarify uncertainties (asterisks, dashes, dissenting calls ..) in a circumscribed set of aligned readings.
Each such set of aligned readings, however, only covers a small part of one chromosome, since complete chromosomes are much too long to pass gel electrophoresis in a single experiment. Therefore, partial solutions have to be attached to each other to form contigs. Contig construction is another task that an expert may be in charge of. Yet another is to separate human base sequences from vector, i.e. virus base sequences etc. All these tasks finally use elementary inferences that can be drawn from intensities of the colours of the four bases. That is why the model of interpretation of those sensory data has to be highly general and reusable. On the other hand, different tasks also combine different nonsensory classification and manipulation methods.
For several reasons we will concentrate in this article on the task of clarifying uncertainties in a set of aligned readings. One reason is that this task creates the highest workload for experts in genome analysis. Automization of this task provides the highest increase in productivity. A second reason is that several effects can be easily explained when modeling this task.
Given a certain task, data are sequentially inspected to localize regions that require rework. Localization is based on symbolic data alone, i.e. on single or aligned sequences of base letters and special symbols. Localization is easy. It can always be made using a single column and always draws upon a deviation from consensus based upon unequivocal readings in all rows.
Whenever a site of deviation has been found, an interesting mixture of problem solving methods is applied. Generally, the problem that causes the uncertainty must first be identified and then removed. Identification means to formulate a hypothesis about which symbol or symbols in a column or sequence of columns are false and which are true. One symbolic appearance of a problem may be compatible with several causes. E.g. a column with two T´s and five asterisks may be a five fold undercall and end up with a T consensus - if the two T´s are very well supported by the sensory data of their readings and T finds at least minor support in the other readings as well. Or it may be a two fold overcall and end up as nothing, if the support for the two T´s by the sensory data is only minor and none of the five asterisks provides additional support.
Side note: The knowledge modelling process The present manual solution for the problem of finalizing base sequencing is conducted by experts at computerized workplaces (UNIX workstations). The more difficult the problem the more does the solution draw upon detailed knowledge of the biochemistry of substances involved, genetics of vectors and inserts etc. Quite some problems can, however, be solved after being taught the major principles and prevailing rules. In other works: Given some background in biology and chemistry - as e.g. taught in undergraduate courses in medical informatics in Germany - and some computer literacy, the routine work of base editing can be learned within weeks by being apprenticed in a lab of molecular genetics.
Therefore, in consent with the project partner we chose a mixture of an ethnographic approach (Meyer et.al., 1989), classical knowledge elicitation techniques, and supervision.
Concretely, a junior knowledge engineer, endowed with basic capabilities in knowledge modelling (a la KADS) and knowledge elicitation, became an apprentice in the Instiute for Molecular Biology in Jena, the largest institution in central Europe involved in the human genome project. He had the opportunity to be taught by experts of different levels and - as it turned out - of different styles of editing base sequences. His suggestions to systematically probe the experts were complied with. In other words, he found excellent work conditions to learn the job through the perspective of the experts. Needless to say that there was no manual or guideline available, because most of the nowadays experts had grown into the job they were now doing and had personally collected their experiences and had each founded them on their individual implicit academic and professional knowledge.
In addition, a senior knowledge engineer experienced in the development of knowledge elicitation methods, in formalizing KADS, and in theoretical foundations of knowledge modelling, served as a supervisor to the junior knowledge engineer. He made sure that structures emerging during the period of apprenticeship, were made explicit rather than becoming internalized and implicit before they could be grabbed.
In combination, a structure evolved that maps the intrinsic properties of the subject matter of molecular biology and the processes involved rather than idiosynchracies of individual editors. To make this structure apparent, we first need to specify the terminology which is at the base of the modelling.
A principle that turned out to provide excellent guidance in introducing appropriate terms was to distinguish between
theoretically: a column where the consensus correctly determines
the true base
pragmatically: a column with identical base symbols in all its
non-blank rows
hypothetically: a column with identical symbols in all its non-blank
rows
deviation from theoretically correct call
symbol that coincides with deviating symbols in its column; deviating symbols may be differing base letters, special symbols in places of bases, or vice versa.
leftmost column where a fault indicator is localized.
column where a fault indicator is localized.
![\includegraphics[width=9.5cm]{symbol.eps}](img6.gif) |
To understand this terminology requires some additional structuring derived from properties of the control structure of the problem solving method. By convention, its inference proceeds sequentially through a fragment in one direction, say from left to right. Since the average share of columns containing fault indicators is below 10%, it can be assumed for the majority of cases that a problem column is preceded by several pragmatically correct columns. Definitely, it will be assumed that the column immediately left of the primary fault column is pragmatically correct. It should be noted that a pragmatically correct column is one where all processes which are based on several biological specimens have ended up in calling the same base. In that case it is highly likely that the column is also theoretically correct. At least is this the working hypothesis of all human experts in editing: never doubt a column where symbols in all non blank rows are letters and are equal.
set of columns right of and including a primary fault column, each of which is itself a fault column. Theoretically a fault region may be indefinitely long. Practically, due to the low share of faults in general and properties of the alignment algorithm they rarely are wider than 3-4 columns.
assumption that a certain symbol in a fault column is faulty.
An atomic fault hypothesis is characterized by
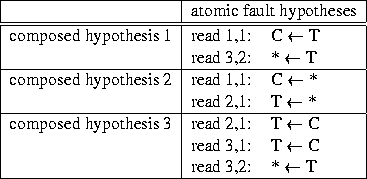 |
Unfortunately we may have both single and multiple faults in the base calling itself and we may have propagating effects of single and multiple faults. We speak of single resp. multiple faults as long as one resp. more than one individual reading fails to call the base correctly. E.g. in figure 3 the C in row 1, column 3 may be a singly misread T. Such cases are covered by atomic fault hypotheses. Or the T's below the C may be multiply misread C's. These two misreads would form a composite fault hypothesis. Both faults have in common that they result from base calling alone and affect one column only. Faults may have a different structure when they result from the process that follows base calling and tries to parallelize different readings (`alignment'). Alignment may require to insert the asterisk symbol in regions where some of the reads display more symbols than others. An inserted asterisk in a read with one fewer symbol manifests the assumption that a base has remained uncovered and will be recovered once attentions has been drawn to the site by the asterisk fault indicator. In contrast to the above single or multiple faults in base calling now more than one column is affected. We speak of fault propagation, when the attempt of the alignment process to compensate for one fault goes wrong. In the example the assumption that a base has remained uncovered may be wrong. Rather the other read which displays more symbols may show a base which truly is not there. All these considerations are to reveal the structure behind composite fault hypotheses. Each composite fault hypothesis can be understood as the sequence of operations that undo one possible single fault, multiple fault, or fault propagation. They generate a constellation of base letters1 that would be present if the error had not been made. Except for very simple cases more than one fault hypothesis is suited to explain the appearance of the fault region. Some hypotheses are more, others are less likely. Likelihood decreases with number and diversity of atomic fault hypotheses involved.
It now becomes obvious that the problem solving method centers around sophisticated generate inferences, whose realizations heavily draw upon structural properties of the domain.
It would go beyond the scope of this article to discuss every detail of the inference structure. But several details will be explained because they illustrate the concept of scalability underlying our approach. We will proceed by relating the roles to concrete domain structures and choices that we have in the domain without changing the inference structure.
The case description consists of a column in the parallelized set of readings, the columns to the right of that column, and the gel eletrophoresis data right of and including the columns2. Different parts ot the case descriptions are used at different stages of the inference. Since a scalable part of the cases is passed to the expert rather than undergoing all inferences, only those few abstractions are done right away, which are required before the decision to treat or to pass the case. Others are only carried out on demand later for those cases that are treated by the system. Abstraction of the primary fault column has to be done immediately, because based on its appearance the decision is made whether to accept or reject (and pass to the expert) the case.
Abstractions at this stage are minor. As an example, for the inference process it does not care in which row(s) certain deviating symbol(s) is/are found. It is sufficient to know the number and type of deviations. Therefore all distributions of a certain number of deviating symbols across rows can be regarded as equivalent. The abstractions applied here map different distributions of patterns across rows upon a common prototypical reference pattern.
Abstract case descriptions are then matched against selection criteria
to accept a case for automatic treatment or to pass it to the expert. The
selection criteria are among the most effective means to scale the system
up or down. They may be formulated as narrow as to accept only cases with
asterisk in the consensus. Or they may just be left away, passing everything
to the automatic system. Of course this "filter" has to be in accordance
with the sophisticatioin of the domain model which is subsequently applied
to solve the cases that have not been passed to the human expert.
![\includegraphics[width=11cm]{inference.eps}](img8.gif) |
Obviously the two decision classes are to accept or reject for automatic treatment. For those cases accepted, the abstraction of the case description now has to span the fault region. The system description introduced just before can be reused here. The fault generation model used for the generate comprises atomic fault hypotheses which can be visualized as rewrite rules for replacing a symbol hypothetized to be faulty by a symbol hypothetized to be true or less faulty. The former means that a letter replaces a symbol. The latter relates to those situations where a dash or asterisk replaces a letter. Epistemologically this means that the strong statement "presence of a specific nucleotid" is replaced by the weaker statement: "presence of some unknown nucleotid" or "assumption of presence of some nucleotid". Scalability comes into play by including fewer or more rewrite rules as base repertoire in the system description. Obviously a system description that includes more rules has the principle capacity to solve more cases. Scalability also comes into play in the definition of the breadth of the fault region achieved by concatenating atomic fault hypotheses. The breadth of the fault region can be set to a definite limit (e.g. primary fault column plus three columns to the right) or can be kept floating according to the column extension of the most extended fault description generated from the abstracted data using the fault generation model.
The fault generation model comprises atomic fault hypotheses and the mechanisms to integrate them into composite fault hypotheses. The respective set of hypotheses and mechanisms is another means of scaling the system. Simple variants may only include single or multiple fault hypotheses and leave away fault propagation hypotheses. In the case of complex fault models with their reach of potentially infinitely many columns interference with the inferences determining the breadth of the fault region, Experiences have to be collected for a fine tuning of these scaling parameters.
In most cases the generate leads to more than one fault description. In case of more than one it suggests itself to establish a ranking among the fault descriptions. This allows to start the subsequent steps first with the most likely fault descriptions. Likelihood is not necessarily a purely statistical measure. Some criteria of the plausibility of a fault description can be derived from their generation process. E.g. fault generation models that require many atomic fault hypotheses are assumed to be less likely than ones with only few atominc fault hypotheses. Fault likelyhood models may also vary with the task under work. Tasks that need to deal with fragments near the (less reliable) end of a read, may require other fault likelihoods than tasks that deal with high quality middle parts of reads.
Given a fault preference list the abstracted fault descriptions can be matched one by one in descending order of likelihood against case descriptions.
It is important to note that so far all inferences have been based upon symbolic information, namely letters for bases and a few special symbols. System description 2 now incorporates sensory information from the original gel eletrophoresis data. The abstraction that takes places is from real valued curves to features of the curve shape (peak height, integral, slope etc.) It goes beyond the scope of this text to provide much detail about this part of domain structure and inferences. However, the reader should have in mind that a potential in scaling up is to take global parameters of individual curves (average energy, known limitations to display clear peaks for certain sequences ...) into account. These, again, may vary for different techniques of staining bases. Therefore, the abstract inferences are general and reusable. But the domain structures may have to be exchange when the specimens have undergone a different experimental protocol.
The decision class resulting after comparing a case description that incorporates symbolic and sensory information with the respective system description 4 is the degree to which the fault description under study explains both composite fault hypothesis and gel electrophoresis curves. If the degree is sufficient the composite fault hypothesis is accepted and the respective repair - i.e. overwriting one or more symbols - is initiated. For some fault regions none of the composite fault hypotheses may achieve a sufficient degree of explanatory coverage. Then a primary fault column that had a priori been accepted for automatic processing may a posteriori be rejected again and then passed to the human expert.
Those cases that reach the repair inference undergo a definitive rewriting of some symbols. There is not much difference between this and the fault generation model. However, conceptually this now is a factual action whereas the generation of a fault description is a tentative temporal suggestion. Furthermore, some fault generation mechanism that involve asterisks have to be done differently in the generate and the repair inferences.
Finally the abstract case description has to undergo the reverse inference of the first abstract in order to map abstract row etc structures into concrete rows of a given fragment. This is achieved by the final specify inference.
If the system is competent to handle a substantial proportion of the edit problems the automated editing will be evaluated by comparing the time needed for computer-assisted editing and the time needed for conventional editing. This value is a meassure for the effect of the system and its ability to achieve the intended aim.
Measuring the correctness of the system is easier than evaluating the effect of the solution but both have to take different personal editing styles into account (e.g. more or less conservative).
![[*]](cross_ref_motif.gif) .
The overall project requirements demand that scaling up is done conservatively.
Thorough quality assurance of stages that attempt to cover more difficult
cases or to apply less clearly understood knowledge has to be done to guarantee
the very low amount of false positive (repair) actions.
.
The overall project requirements demand that scaling up is done conservatively.
Thorough quality assurance of stages that attempt to cover more difficult
cases or to apply less clearly understood knowledge has to be done to guarantee
the very low amount of false positive (repair) actions.
However, the amount of manual work along the lines of this article that is required until full decyphling of the human genetic prodigy has been estimated to amount to much more than 100 person years. If his estimate is correct, a very low scaling first solution, which covers about 25% of the most common and not too hard cases, already saves 25 person years of work compared to a development cost of much less than half a person year. Therefore, the presented approach is not only structurally interesting and challenging but also of high economic value.
The organisational environment of genome sequencing lends itself for an ethnographic approach with supervision. In genome sequencing the knowledge engineer can more truely become a junior team member than in many other situations. Compared to many tasks in medical domains, control of complex industrial processes etc., the risk of errors in the apprentice phase comes close to zero. The knowledge engineer in his role as a junior team member among biochemists and molecular biologists can just try and apply what he has learnt the day before. The senior team member can check, discuss and, if necessary, revise the early attempts of the knowledge engineer. Therefore, growing from an apprentice to a junior team member role is quite natural in the setting of genome sequencing. As a matter of fact, new human members of the team didn't and don't learn differently than the knowledge engineer. This makes obvious the need for supervision. Because the natural way of training an apprentice is to enable him for doing the job rather than enabling him to analyse and make explicit the structures underlying the job. Therefore, a procedure that carefully balanced the normal process of apprenticeship with a "KADS-filtered" supervision allowed to arrive at a detailed and sophisticated model within short time.
The authors thank B. Drescher and M. Platzer for their highly efficient guidance into the subtleties of molecular genetics and base sequencing.
This work is supported by the Bundesministerium für Bildung, Wissenschaft,
Forschung und Technologie by grant number 01 KW 9611.
(Breuker & van der Velde, 1994)
Breuker, J. & van der Velde W. (Eds.)
(1994):
CommonKADS Library for Expertise Modelling.
IOS Press Amsterdam.
(Bürsner & Schmidt, 1995)
Bürsner, S. & Schmidt, G. (1995):
Building views on conceptual models for structuring
domain knowledge.
In: Proc. 9th Banff Knowledge Acquisitions for Knowledge-Based
Systems Workhop,
Department of Computer Science, University of Calgary,
Calgary. SDG Publication.
(Meyer et.al., 1989)
Meyer, M., Picard, R. R., & Ross, J. M. (1989):
The ethnological method of participant observation
as a technique for manual knowledge acquisition.
Proc. 5th AAAI-Workshop on Knowledge Acquisition,
Banff Canada.
(Myers & Weber, 1997)
Myers, E. W. & Weber, J. L. (1997):
Is whole human genome sequencing feasible?, pages
73--90.
In (Suhai, 1997).
(Schmidt & Wetter, 1998)
Schmidt, G. & Wetter, T. (1998):
Using natural language sources in model-based knowledge
acquisition.
Data & Knowledge Engineering. in press.
(Setubal & Meidanis, 1997)
Setubal, J. & Meidanis, J. (1997): Introduction
to computational molecualar biology.
PWS Publishing Company.
(Suhai, 1997)
Suhai, S., (Ed.) (1997): Theoretical and computational
methods in genome research.
New York, Plenum Press.
(van Harmelen & ten Teije, 1998)
van Harmelen, F. & ten Teije, A. (1998):
Characterising problem solving methods by gradual
requirements: overcoming the yes/no distinction.
same volume.
(Wielinga et.al., 1992)
Wielinga, B. J., Schreiber, B. J., & Breuker,
J. A. (1992):
KADS: a modelling approach to knowledge engineering.
Knowledge Acquisition, 4(1):5--54.